




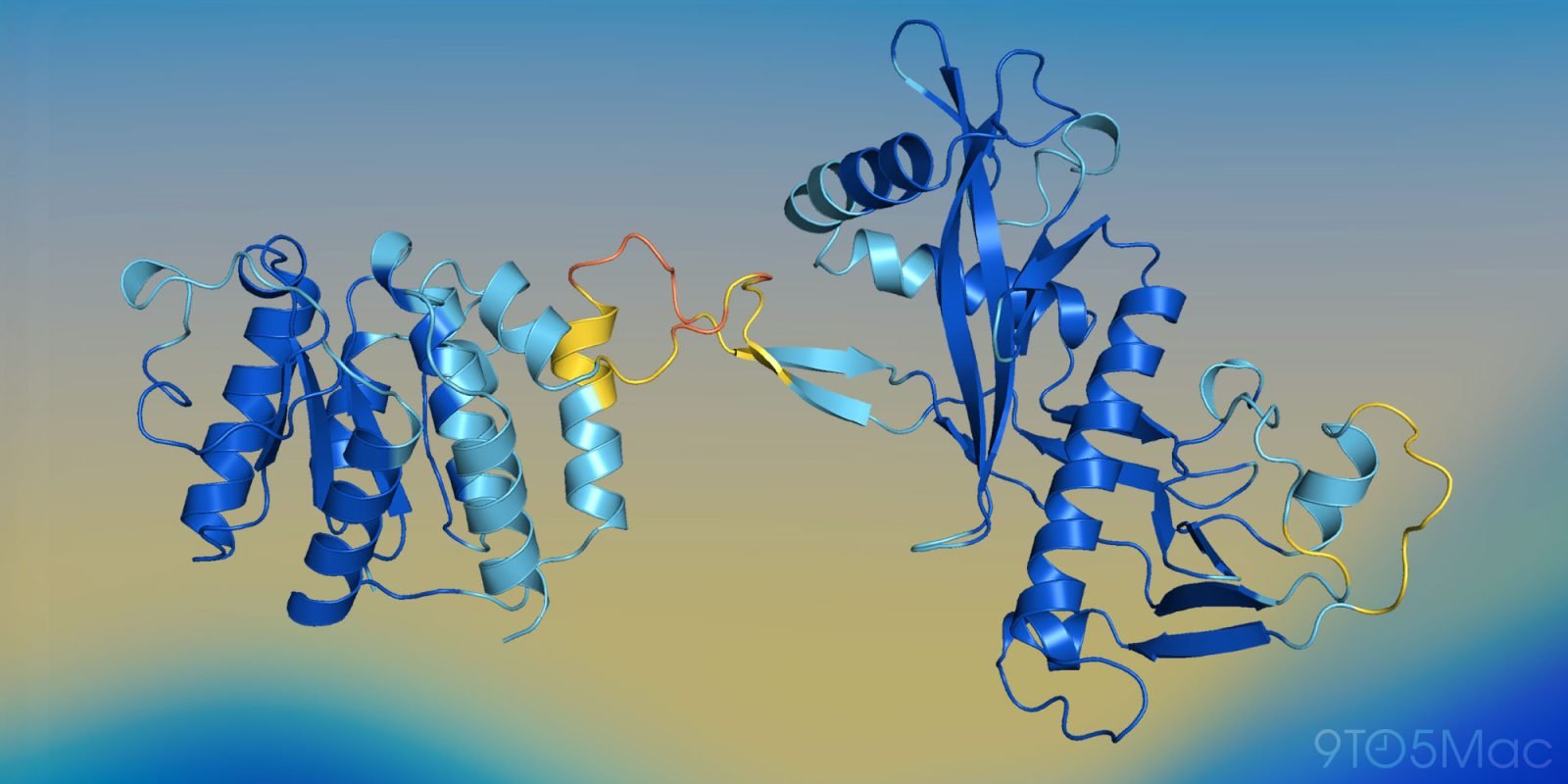
Google DeepMind的AlphaFold堪称奇迹,但计算成本高昂。为此,苹果研究人员开发了一种利用AI预测蛋白质三维结构的新方法,并展现出巨大潜力。以下是详细内容。
AlphaFold是Google DeepMind的革命性AI模型,能够根据氨基酸序列预测蛋白质三维结构。这对开发更有效的药物及全新材料具有重大价值。
数年前,这曾是个极其困难的课题。预测单个蛋白质的三维原子结构往往需要数月甚至数年时间。
但随着AlphaFold、AlphaFold2以及RoseTTAFold、ESMFold等尖端模型的出现,这一过程已缩短至数小时甚至数分钟(取决于硬件配置)。
这些模型虽采用不同方法实现高精度,但普遍需要极高计算成本,且框架结构极为严格。
正如苹果研究人员所述:
“现有蛋白质折叠模型(如AlphaFold2和RoseTTAFold)通过精心设计的架构实现突破性精度,这些架构整合了计算密集的特定领域设计,包括氨基酸序列多重比对(MSA)、配对表征和三角更新等。这些设计本质上是将当前对结构生成过程的理解硬编码至模型中,而非让模型直接从数据中学习——后者可能带来多方面的优势。”
苹果SimpleFold的突破
该创新模型摒弃了”MSA、配对交互图、三角更新或等变几何模块”,转而采用2023年提出的流匹配模型——这种在文生图、文生3D领域已获验证的技术。
简言之,流匹配模型是扩散模型的进化版。不同于传统扩散模型逐步去除初始图像噪声,它能学习更平滑的路径,将随机噪声一次性转化为完整图像。
由于跳过多步降噪过程,该方法计算成本更低且生成速度更快。
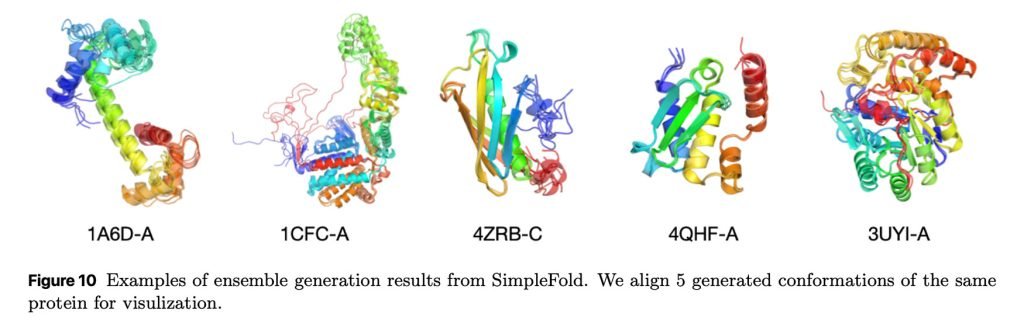
苹果团队以1亿、3.6亿、7亿、11亿、16亿及30亿参数规模训练SimpleFold,并在CAMEO22和CASP14两大蛋白质结构预测基准测试中验证其泛化性、鲁棒性及原子级精度。
结果令人振奋:
“尽管设计简洁,SimpleFold性能媲美基线模型。在两项测试中,其表现始终优于同样采用流匹配的ESMFlow模型。在CAMEO22中,SimpleFold与顶尖折叠模型(如ESMFold、RoseTTAFold2和AlphaFold2)结果相当。值得注意的是,在不使用昂贵启发式三角注意力和MSA的情况下,SimpleFold在多数指标上达到RoseTTAFold2/AlphaFold2 95%以上的性能。”
以及
“为完整呈现,我们展示了不同规模SimpleFold的结果。最小的1亿参数模型在训练和推理效率优势下仍具竞争力,在CAMEO22上达到ESMFold 90%以上的性能,证明通用架构模块构建折叠模型的可行性。”
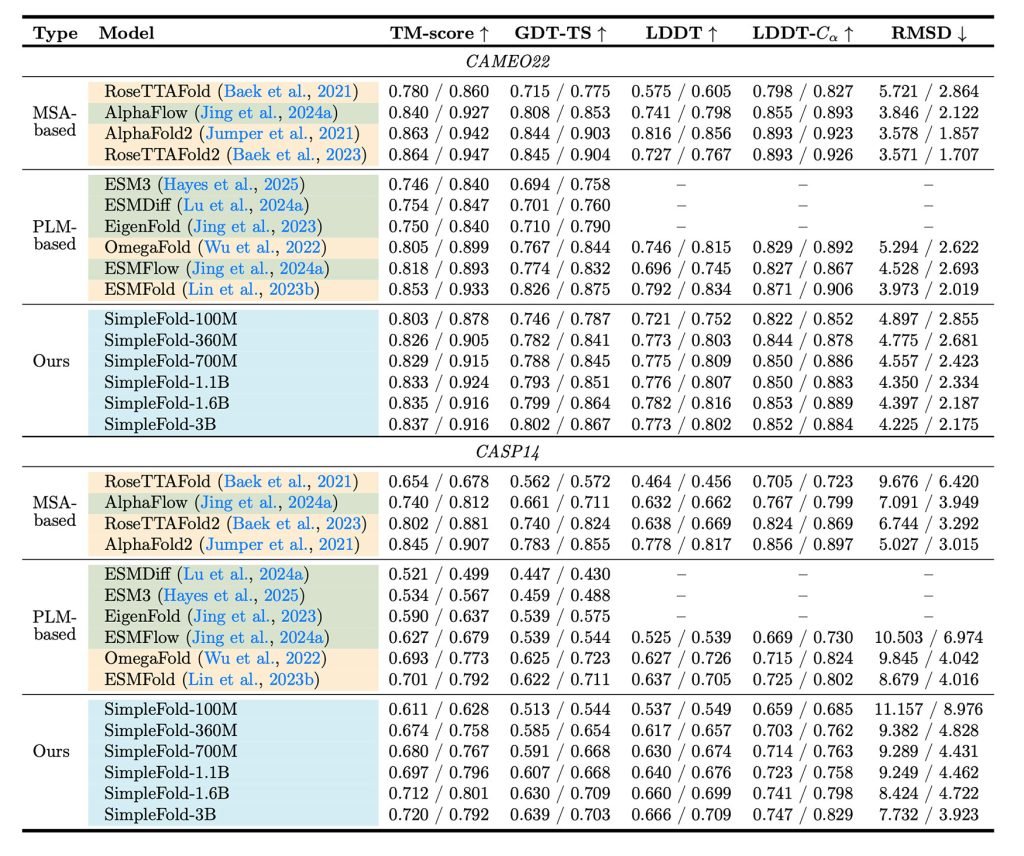
研究还发现性能随模型规模提升而增强,表明更大模型配合更多训练数据可显著提升折叠性能,尤其在最具挑战性的测试中。
团队强调SimpleFold仅是第一步,希望其”能推动学界构建更高效强大的蛋白质生成模型”。
完整论文可参阅arXiv。











